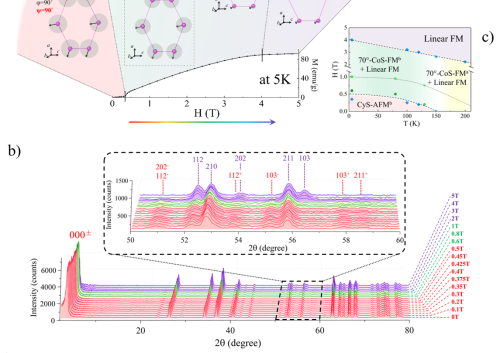
当样品在X射线衍射(XRD)实验过程中发荧光时,定量结果中产生的误差可能使实验毫无意义。在用铜X射线管对含铁的样品进行测量时,荧光尤其常见。
许多人吹捧使用衍射光束单色仪或能量鉴别检测器来净化信号,但样品荧光表明将存在微吸收效应,而微吸收几乎总是会导致结果误差。
当来自源的入射X射线激发样品的原子,从而导致它们发出自己的特征X射线时,就会发生荧光。

当源K-α辐射能量仅略大于样品的特征X射线时,此效应最为明显。不幸的是,最大的效果与最大的困难在于将荧光x射线与源x射线区分开来,因为它们的能量差别不大。由于具有相似能量的X射线如此之大的强度,衍射图谱倾向于具有大大提高的背景。
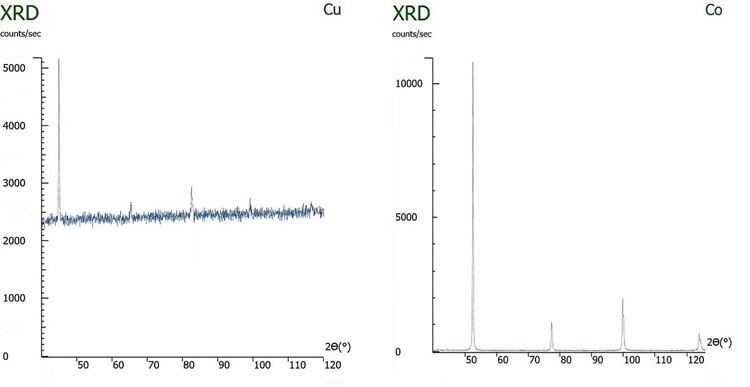
衍射光束单色仪或高能分辨率检测器可以区分和排除样品荧光,从而大大降低了背景,但它们并没有减轻微吸收的影响。当源辐射最有效地刺激样品中的荧光时,样品对源辐射的吸收将最有效。如果样品具有多种成分(在进行定量相分析时会如此)且它们的X射线吸收常数不同,则微吸收极有可能使表观组成发生严重偏差。
样品中微吸收的重要性取决于其组成相的粒径(D)和线性衰减系数(μ)。线性衰减系数表示每单位厚度将被吸收(或衰减)的入射X射线束的比例。根据Brindley标准(不更正)),为了通过XRD进行可靠的定量分析,μD值应小于0.01。另一方面,如果μD大于0.01,则大部分X射线将被材料吸收而不是被衍射,从而导致该分量在结果中的表示不足。重要的是要记住,如果元素或化合物的线性衰减系数之间存在显着差异,则在具有多相的混合物中,衍射峰的相对强度可能会根据粒径而变化。
尽管Brindley标准可以帮助确定样品是否会表现出微吸收效应,但这种方法仍存在挑战。首先,通常难以量化粒径,因此无法知道μD的值。许多实验室只能使用筛子,因此没有精确的方法来测量粒度。其次,即使可以通过研磨识别和改变粒径,某些材料也需要极小的粒径才能使μD低于0.01。例如,可能需要将颗粒研磨成小于一微米的颗粒。在这种情况下,不太可能使用必要的设备来减小粒径并验证减小的程度,从而导致μD值大于0.01。
在固体样品上,微吸收的问题甚至更难缓解,因为晶粒大小将决定微晶的大小。因此,如果样品的晶粒不是非常细,并且所有相的μ都不相似,则该材料将不符合Brindley标准,因此实际上在任何定量分析中都会出现误差。
为了分析荧光和微吸收的影响,将铁和镍以等原子比混合。在经过一定的研磨时间后,用铜和钴源对样品进行了测试。如下图所示,当使用铜X射线源(在铁中诱发大量荧光)进行测试时,铁-镍混合物的成分被错误地表示:未经研磨,所示镍的原子分数约为70%,而铁的原子分数为30%。当样品研磨一个小时后,测得的原子组成与实际组成非常接近,表观镍组成约为55%,铁约为45%。但是,当X射线源从铜切换为钴时,铁中的荧光较少,
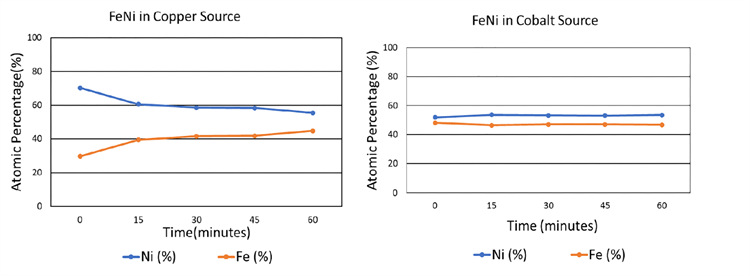
根据这些结果,很明显,选择兼容的X射线管可以是减少微吸收效应的高效解决方案。当(1)背景噪声高,(2)被测组件的线性衰减系数非常不同,(3)被测组件的线性衰减系数非常高,(4)组件的线性衰减系数时,这一点尤其重要未知,(5)观察到丢失的峰,或(6)使用无标准的分析方法(例如Rietveld)。
应该强调的是,高背景水平不是问题。相反,它们表明存在较大的μ(进而为μD)和微吸收引起的误差的可能性。通过单色化或能量歧视来减少背景不会改善由微吸收引起的不准确性。因此,唯一有保证的解决方案是更换X射线管。